- 2018-05-21 10:40
- 作者:中信建投证券 贺菊颖
- 来源:乐晴智库精选
国际巨头重金加码细胞治疗
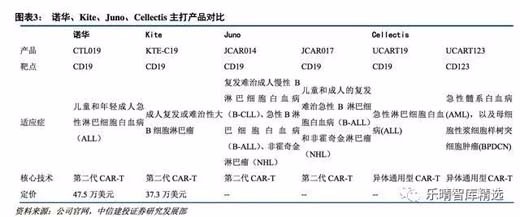
国际巨头纷纷布局CAR-T业务,诺华、吉列德(KitePharma)、新基(JunoTherapeutics)三巨头领跑。
自从宾夕法尼亚大学CarlHJune教授实现CAR-T细胞治疗后,国际上已有多家公司开展CAR-T疗法的进一步探索。
最早进行CAR-T细胞治疗开发的三家公司为诺华、KitePharma以及JunoTherapeutics,KitePharma被吉列德以119亿美元收购,Juno被新基以90亿美元收购剩余90%股权,交易完成后,Juno成为新基全资子公司。
2018年3月,由凯特制药(KitePharma)两位前高管创办的AllogeneTherapeutics获得3亿美元融资,创下了初创公司的融资估值和融资速度的记录。
诺华制药(Novartis)作为进入CAR-T领域的较早的制药公司,与美国宾夕法尼亚大学的合作,已经拥有领先的CAR-T研发管线,成功入驻第一梯队。
2014年7月根据新药临床试验申请(IND),将CTL019用于治疗儿童和成人复发或难治性的急性淋巴细胞白血病(ALL)患者;2015年4月在全球范围内启动了CTL019治疗ALL的2期临床试验,12月中期临床数据显示接受CTL019治疗后,完全缓解率达到了93%。
2017年8月CTL019(Kymriah)获得FDA批准上市,成为人类历史上首个上市CAR-T治疗产品。
凯特制药(KitePharma)是CAR-T治疗新星,已被吉列德以119亿美元收购。2016年9月Kite公布了KTE-C19的二期临床研究中期数据,弥漫性大B细胞淋巴瘤患者总体反应率达到76%,完全缓解率达到47%;转移滤泡性淋巴瘤患者总体反应率达到91%,完全缓解率达到73%。
同年12月KitePharma已就其CAR-T疗法KTE-C19向FDA提交上市注册申请,针对的适应症为不适合自体干细胞移植的复发/难治侵袭性B细胞非霍奇金淋巴瘤(NHL)。2017年10月其产品KTE-C19(Yescarta)获得FDA批准上市。
JunoTherapeutics,已被新基公司(Celgene)以90亿美元收购,Juno与纪念斯隆-凯特林癌症研究中心(MemorialSloanKetteringCancerCenter)、弗雷德·哈金森癌症研究中心以及西雅图儿童医院(SeattleChildren'sHospital)三家研究中心合作,集结了领域内顶尖的专家。
尽管首个临床产品JCAR015因严重副作用而终止,另外两个研发项目JCAR014和JCAR017临床结果较好,为Juno带来了曙光;JCAR017在临床数据上的表现比KTE-C19更优秀,JCAR017或将成为非霍奇金淋巴瘤患者的新选择,有成为Best-in-Class产品的潜质。
目前经过改进的JCAR017在治疗复发或难治的B细胞非霍奇金淋巴瘤中,有43例可评估案例,3个月的总体反应率ORR为80%,完全缓解率CR为73%,预计Juno将于今年下半年提交CAR-T注册申请,最快将于年内获批。
自2012年诺华和CarlJune教授合作开发CAR-T疗法以来,美国多家大型药企加入了CAR-T研发竞争的行列,纷纷与CAR-T研发公司合作,Juno、Kite、Cellectis、Bluebird、南京传奇等公司目前均与跨国药企巨头签署了研发或销售合作协议。
细胞治疗仍存两大预期差
细胞治疗在二级市场已获得较高关注度,但我们认为仍存两大预期差,第一,细胞疗法的商业价值仍未被市场充分认识;第二细胞疗法的商业化进程可能被低估。
股价并未完全反应细胞治疗的商业潜力:全球细胞治疗龙头KitePharma市值120亿美元,JunoTherapeutics市值100亿美元,Bluebird市值85亿美元,我国细胞治疗龙头公司金斯瑞生物科技布局与Bluebird、Juno相近,LCAR-B38M-CAR-T在多发性骨髓瘤适应症研发进度第一。
临床表现上,2017年ASCO大会上,非完全可比口径,南京传奇的LCAR-B38M录得更高完全缓解率(73%vs27%),首批25位治疗患者已治疗超过1年,其中20位仍维持完全缓解状态,具有成为First-in-Class及Best-in-Class产品的潜力,市值仅445亿港元(对应57亿美元)。
市场主流观点认为细胞治疗只能用于复发难治血液肿瘤,市场空间相对较小,我们认为随着研发推进,细胞治疗将用于非复发难治血液肿瘤及实体瘤,有望把肿瘤变成慢性病,市场空间巨大。
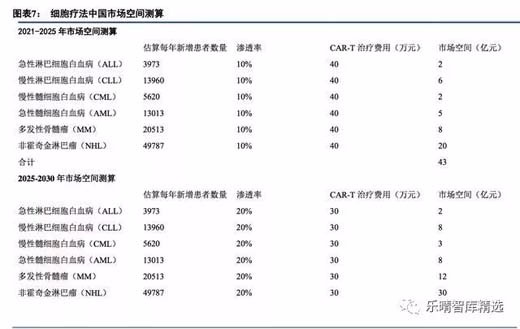

市场主流观点认为细胞治疗在我国商业化尚早,还没到贡献利润的时候,我们认为龙头公司有技术积累,有商业化能力,药审鼓励创新和急需用药,商业化进程有望超出市场预期,最快有望在2019年初实现商业化。
市场主流观点对细胞治疗研发公司进行推荐,我们认为细胞治疗研发服务商也同样值得关注,细胞治疗产品安全性评价市场空间在15-21亿之间,且随着新增靶点推进,市场需求会持续增长,重点推荐细胞治疗安全性评价领域相关标的。
此外,细胞治疗产品慢病毒载体市场空间在10亿元左右,且慢病毒载体评价周期长、壁垒高,随着细胞治疗产品的上市,有望形成类似独家CMO供应模式,建议关注慢病毒载体供应商。
预期差之一:商业价值被低估
不仅仅是“复发难治”肿瘤
目前已获批上市的Kymriah和Yescarta适应症均为复发难治血液肿瘤,但这是出于伦理学的要求和注册策略的选择。
我们认为,这并不意味着细胞治疗必须用于治疗复发难治的血液肿瘤,而是有望向前推移。
以自体造血干细胞移植治疗多发性骨髓瘤为例,目前无进展生存期为3-5年,以细胞治疗产品临床阶段的表现,有可能替代自体造血肝细胞移植。
预计临床试验设置为试验组(细胞治疗产品)、对照组(自体造血干细胞移植),比较二组无进展生存期,假设细胞治疗获得更好的临床数据,细胞治疗的市场将从复发难治血液肿瘤拓展到非复发难治血液肿瘤。
从血液肿瘤到实体瘤
虽然细胞治疗在血液肿瘤上已经证明疗效,但是在实体瘤上的应用前景则存在分歧,我们认为细胞治疗下一个重要目标就是攻克实体瘤,目前细胞治疗用于实体瘤已经在部分癌种和靶点上取得让人激动的成果。
2015年11月,美国肿瘤免疫学年会(SITC2015),NY-ESO-1靶点TCR-T疗法的用于滑膜肉瘤的临床试验数据(NCT01343043)发表,总体应答率达到50%,而且T细胞在患者体内持续存活。
2018年2月,ScienceTranslationalMedicine报道,斯坦福大学医学院的研究人员将两种免疫刺激剂(OX40抗体以及CpG的寡核苷酸短DNA片段-TLR9)直接注射到90只小鼠的实体瘤中,有效消除87只小鼠体内所有的癌症痕迹,另外3只肿瘤复发,在经过第二次治疗后都再次消退。
2018年3月,Nature杂志发表一组来自日本的科学家团队的研究,通过将IL-17以及CCL-19基因转入CAR-T细胞制备出能够有效杀伤肿瘤的“超级CAR-T细胞”,在小鼠实验中效果相当惊喜,荷瘤小鼠生存率达到100%,相比之下,采用常规CAR-T疗法的肿瘤小鼠生存率只有30%左右。
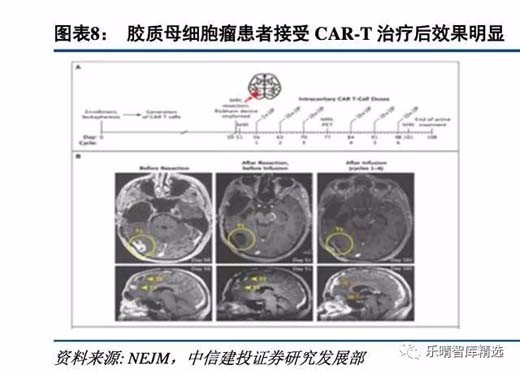
国内企业在实体瘤治疗大潮中激流勇进
科济生物2017年公布了全球首个针对GPC3靶点的、使用细胞治疗难治复发肝癌的I期临床试验结果。13名CAR-T治疗的患难治复发的肝细胞癌患者均耐受良好。
随后将开展全球1b/2期临床,在5名可进行疗效评估的患者中,1名患者出现部分缓解,2名疾病稳定,2名患者疾病进展(一名在治疗12月后死亡)。
4名病人生存期分别超过14个月、20个月、14个月和10个月。2017年12月28日,CAR-T-GPC3获得CFDA的受理,是国内首个用于实体瘤临床试验的CAR-T。
此外,科济生物还布局针对胶母细胞瘤的EGFR/EGFRvIII双靶点CAR-T的临床试验,针对胃癌、胰腺癌的Claudin18.2靶点治疗的CAR-T的临床试验。
尽管当前还没有实体瘤细胞治疗产品进入大规模人体试验,但我们对技术进步持有乐观态度,随着新型高特异性靶点的发现及基因编辑技术的快速进步,实体瘤攻克将成为可能。
纳入医保,解决患者沉重负担
尽管Kymriah和Yescarta已获准上市,但由于高昂的治疗费用和不确定的医保支付政策,目前大量美国患者目前有大量患者在排队等待,除非签订文件表示愿意在医保不予支付时承担全部治疗费用。
根据BiopharmaDive信息,美国医疗保险巨头CMS在4月初将细胞疗法Kymriah、Yescarta纳入其医保B计划,并将分别支付上限50万美元、40万美元。我们认为CMS做法符合药物经济学逻辑,原因如下:
- Kymriah用于治疗复发急性淋巴细胞性白血病(ALL),该疾病复发患者一般需要异体骨髓移植治疗,费用在50-80万美元,且后续几年维持用药仍有高额费用;Yescarta用于治疗弥漫性大B细胞淋巴瘤,需要造血干细胞移植的费用也在30万美元以上;
- Kymriah一次治疗费用47.5万美元,Yescarta一次治疗费用37.5万美元,且根据Kymriah数据,60%患者在至少14个月后仍维持治愈状态,即具有一次治愈的可能,长期看有一定经济性。
- 细胞疗法从定价上基本与骨髓移植可比,远期费用上看,甚至可能低于骨髓移植,假设细胞疗法的使用往前推移替代骨髓移植,则经济性将更显著。
中国骨髓移植费用一般在20万元左右,移殖前后用药成本大概在30万元左右,考虑医保报销后,患者自付部分约为10万元,作为有一次性治愈可能的疗法,细胞治疗定价30-50万并不算很贵,我们对细胞疗法纳入医保持乐观态度。
降低细胞疗法生产成本
T细胞肿瘤免疫疗法通过从患者或者捐献者外周血分离获得特定亚群的T细胞,进一步激活、基因修饰获得CAR-T细胞,扩增培养从而到达治疗级别的细胞数,再经过洗涤浓缩回输患者体内进行治疗。
CAR-T制作过程的无菌操作、纯度、制备结果的安全性等质量控制对后期的疗效有着重要影响,所以确保生产过程的标准化和自动化,做到药效和风险可控是首要任务。
从分离收集T细胞,到激活修饰再到扩增细胞、回输病人体内是复杂的流程,传统人工制备面临极大挑战:
- 第一是环境挑战,制备过程污染的可能性很大;
- 第二是人为差异,每个操作人员的偏好会导致产品参差不齐,产生差异性;
- 第三是作为高度个性化产品,每个患者即一个生产批次,与传统制药工艺有较大差异;
- 第四,血液肿瘤患者病情进展极快,需要尽量缩短制备时间及时回输,并提高成功率。
因此,CAR-T生产过程的标准化、自动化以及安全性、无菌性、纯度等质量控制是客观评价缓解率的先决条件。
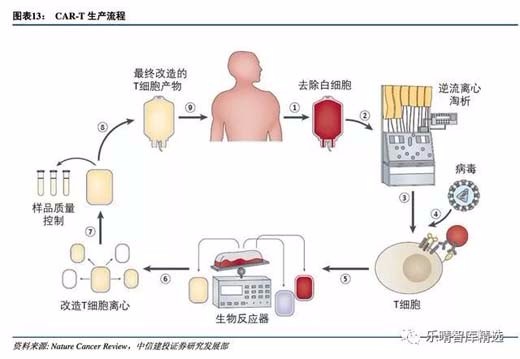
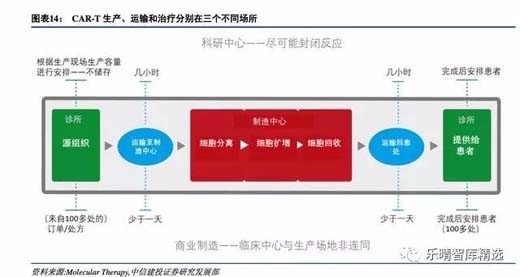
化解高成本压力:从自动化和慢病毒开始
CAR-T生产成本高达30万美元,其原因在于三点:
- CAR-T作为高度个性化的药品,一个患者对应一个批次,制备操作需要多名高端技术人员进行无菌精准操作,人力成本极高;
- 为了保证洁净度、纯度和制备结果的安全性,需要建设大面积专业化无菌操作区间,专业化的清洁人员需要定期进行维护,运营成本极高;
- 转染CAR使用的慢病毒的稳定性和安全性对CAR-T产品制备的质量和使用安全性影响很大,优质的慢病毒采购成本高达5-10万元,而自制慢病毒又难以解决稳定性的问题。
诺华旗下CarlJune团队的CTL-019疗法中宾夕法尼亚大学的临床细胞和疫苗生产中心(Clinicalcellandvaccineproductionfacility),符合FDA-GMP标准,所有GMP车间都是由受过专业培训的操作人员按照既定程序,各种关键设备严格管控,实现整个生产过程的污染防止。
随着更多CAR-T产品上市,未来有更多病人从中受益,面对如此庞大的市场需求,CAR-T细胞制备过程的全自动化、标准化将是各大细胞治疗公司重点关注的领域。

自动化设备的使用有利于降低成本
多家CAR-T研发企业已经形成自己的工艺流程,其中最为成熟的是德国Miltenyi公司的全自动平台CliniMACSProdigy,实现病人血液细胞的分选,转染,扩增自动化,避免生产过程中多个设备之间切换造成的人员成本和污染风险。
CliniMACSProdigy是目前全球唯一的CAR-T全自动化制备产品,密闭安全、自动方便、用户友好,不受基础设施的限制,易于扩大CAR-T细胞制备的适用范围;而且全自动提取,批次稳定性有效性很好,远远超过手工制作。

慢病毒载体的本土化,有望进一步降低成本
慢病毒载体是CAR-T细胞治疗产品生产的关键原料,对产品最终质量有着至关重要的影响。慢病毒制备过程复杂(图表19),一般至少需要两周时间,而且需要在超净环境下,并在符合GMP标准的环境下生产,因此成本较高。理想的慢病毒载体应当具有稳定性和安全性。稳定性是指保证在不同地点,不同患者批次CAR-T细胞生产中,获得相对一致的效果。安全性则需要治疗后患者的长期随访跟踪。
考虑到稳定性和安全性的高要求,诺华的CTL019,Kymriah选择OxfordBiomedica公司作为慢病毒载体唯一供应商,签订3-5年的供应合同,预计3年内OxfordBiomedica对诺华销售1亿美元,预计细胞治疗用慢病毒市场在10亿美元以上。
OxfordBioMedica是一家慢病毒载体领域的龙头公司,拥有丰富的肿瘤、中枢神经系统、眼科相关的基因和细胞治疗管线,并与诺华、赛诺菲、GSK等一线药企建立了良好合作关系。
2017年4月,复星医药和美国Kite共同设立的合营公司,致力于在中国商业化细胞治疗产品。
2017年12月,强生旗下杨森制药与金斯瑞生物科技签署了关于多发性骨髓瘤领域的若干产品的开发、制造和商业化的特许协议,杨森公司将帮助金斯瑞建造细胞治疗生产设施。
2018年1月,博生吉安科与德国Miltenyi公司达成合作备忘录,在合肥建设基于CliniMACSProdigy和MACSQuant平台的全自动CAR-T细胞制备工厂,致力于CAR-T疗法各个领域展开深度合作。
药明康德在美国建立的子公司AppTec(药明康德美国)针对CAR-T细胞疗法开展以下服务:1)生产工艺研究,包括工艺优化、平台发展、工艺放大与工艺验证;2)面向临床试验的细胞治疗和基因治疗产品的小批量生产;3)细胞治疗和基因治疗产品的商业化生产;4)相关测试服务。
除此之外,以吉凯基因、和元上海、上海复百奥为代表的我国慢病毒供应商正在崛起,为我国细胞治疗产品提供优质低价的慢病毒载体。
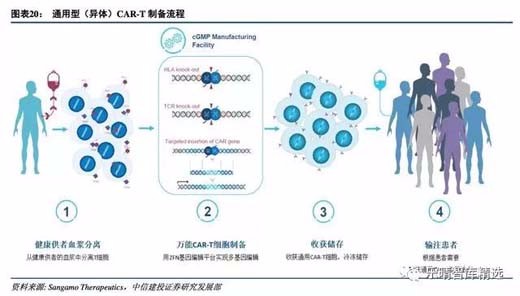
降低成本的终极武器:从个性化到通用型
自体CAR-T细胞治疗是一种个性化的治疗方式,生产成本极高,且由于患者疾病进展较快,自体CAR-T细胞治疗产品生产周期较长,另外,对于婴幼儿患者、老龄患者以及化疗、放疗后的病人,体内T细胞的数量和质量都不能达到CAR-T治疗的要求,部分患者难以及时回输治疗,失去延长生命的机会。
通用型CAR-T的开发排除个体差异,实现规模化生产,从而降低成本,降低治疗费用,缩短回输时间。诺华临床数据显示,自体CART制备的失败率是10%,因此异体CAR-T治疗无疑对这部分病人以及未来的规模化量产由很大的现实意义。

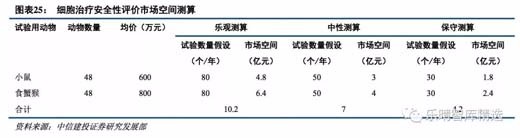
我们认为根据现行的细胞治疗产品安全性评价办法,细胞治疗产品安全性评价市场空间中性测算在 7 亿元 左右,乐观测算假设为小鼠安评每年 80 个,均价 600 万元,食蟹猴安评每年 80 个,均价 800 万,对应市场空 间 10.2 亿元;中性假设为小鼠安评每年 50 个,均价 600 万元,食蟹猴安评每年 50 个,均价 800 万元,市场空 间为 7 亿元;保守测算假设为小鼠安评每年 30 个,均价 600 万元,食蟹猴安评每年 30 个,均价 800 万元,市 场空间为 4.2 亿元。
目前药物安全性评价市场约 10 亿元,细胞疗法研发的繁荣给药物安全性评价带来的弹性巨大。我们预计,随着新靶点的持续开发,实体瘤细胞疗法的研发热情进一步提高,细胞治疗的临床试验数量会 持续增长。
预期差之二:商业化进程有望超预期
我国科研水平及科研临床数处于第一梯队
2017年12月南京传奇生物的CAR-T产品首家获得CDE受理并进入优先审评通道,截至目前CDE受理CAR-T申报数量增至12个,其中4个产品进入优先审评。
2018年1月29日,CDE拟将2017年申报的另外三个CAR-T纳入优先审评通道,反映出CFDA对加快CAR-T疗法审评的积极态度。
预计CAR-T疗法申报均具有纳入优先审评资格。2018年3月13日,南京传奇LCAR-B38MCAR-T细胞自体回输制剂收到CFDA的临床批件,成为国内首个获得临床批件的公司。
从注册临床试验数来看,中国现在是全球最活跃的CAR-T临床试验国家,数量超过北美,且远超欧洲、日本等地。
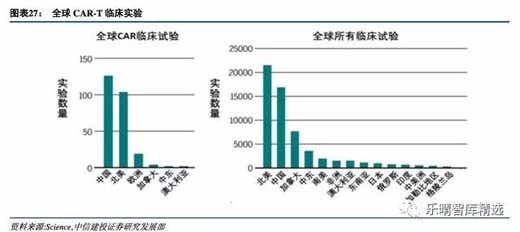
国内政策明朗,药审效率提升,为CAR-T发展锦上添花。
不同于传统的药物,CAR-T是活体细胞,需要在特定场所实施治理;另一方面,CAR-T接受基因工程改造,也属于基因治疗药物。
基于CAR-T特殊性,如何进行监管成为一个难点,国家政策的大力支持,使得CAR-T监管明朗化,药审部门明确CAR-T纳入优先审评,为CAR-T加速上市提供助力。
自2016年12月16日,发布《细胞制品研究与评价技术指导原则》(征求意见稿)以来,细胞治疗政策落地反映国家对医疗创新技术的高度支持和鼓励,将直接利好细胞治疗技术公司。2017年12月CFDA发布了《细胞治疗产品研究与评价技术指导原则(试行)》,国家层面政策逐渐明朗,预计2018年将继续有多家生物技术公司提出细胞治疗技术临床研究申请,产业投资热潮即将来临,2018年有望成为CAR-T投资大年。
中美细胞疗法科研实力相近,我国有望跑出全球核心资产
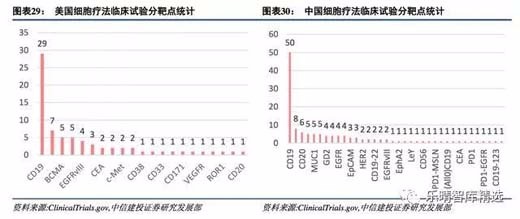
从试验靶点及适应症上看,我国临床靶点种类及适应症也是紧跟美国,二者科研临床水平不相伯仲。
从我国在ClinicalTrials.gov上登记的临床试验看,目前已覆盖43个靶点或靶点组合。相比之下,美国覆盖大约20个靶点或靶点组合。CD19、CD22、BCMA在中美两国均为最热靶点,两国都在EGFRvIII、MSLN、CEA等新兴靶点上有试验,两国均有通用型CAR-T注册试验。
不同之处在于美国的靶点创新更多是基于已有的成熟靶点嫁接于CAR-T,例如VEGFR、HER2、C-Met,中国的创新侧重于重新组合(CD19-22,CD19-PD1等)和全新靶点(CD80、CD86、CD56、CD117等)。
具体看最热的三个靶点CD19、CD22、BCMA:
1)CD19非常成熟,美国已有两个上市品种,我国已有50个注册临床,9个品种申报IND,由于CD19确定性高,这一领域差距只是时间问题;
2)CD22美国临床数7个,我国8个,作为桥接CD19-CAR-T的品种,我国并不落后;
3)BCMA靶点被誉为“下一个最具成药性”的靶点,美国主要是Bluebird公司在推进,KITE2017年下半年启动临床试验,JUNO临床试验处于临床I期招募状态,我国南京传奇公司(金斯瑞生物科技)已进行接近两年时间科研性临床。
南京传奇是拥有全球唯一一个BCMA靶点的双表位CAR结构知识产权,拥有更高的理论结合效率,且在非完全可比口径下录得更高的完全缓解率、复发率,目前传奇公司正与强生公司共同推荐全球范围商业化。
我国有望跑出全球核心资产
在细胞治疗领域,我国与欧美差距很小的,具有一定的历史必然性。小分子新药爆发的时代,是美国的1900年-1970年,我国彼时仍处于战乱和封闭状态,科研实力和制药实力薄弱,错过了发展机遇。
抗体药时代是1980年开始,以利妥昔单抗上市为标志,但是1980年我国改革开放刚开始,工业实力和科研实力仍处于培育中,再次错过发展良机,但是我国在抗体药领域的差距已大幅缩小,如今已经达到快速跟踪全球新进临床靶点的水平。
2005年开始的细胞治疗时代,我们在基础科研上已经逐步靠近欧美水平,我国患者数量庞大,大量海外优秀科学家也在2000年之后回国任教或创业,靶点发现具有复杂性、偶然性的特点,我国细胞治疗布局靶点多,研发投入大,发现重磅靶点概率高。天时地利人和,我国有望在细胞治疗领域弯道超车,跑出全球核心资产。
临床试验及商业化进程有望超预期
CAR-T疗法作为突破性疗法,又兼具临床急需特点,商业化进程有望加速,理论上可申报有条件上市。
传统药物审批需要经过三期临床试验,其内在逻辑在于:临床I期用于安全性评价,临床II期用于剂量选择参考,临床III期用于大规模验证效果。我们认为,细胞治疗作为一种新型治疗药品,临床试验有一定特殊性。
首先,当前细胞治疗临床产品主要适应症为复发难治的血液肿瘤,患者疾病进展很快,难以等待产品上市,其次细胞治疗目前仍是高度个性化产品,剂量和最终产品活性跟患者具体病情进展,身体状态差异,T细胞质量,对免疫因子风暴的耐受程度都有较大关系,难以确定统一的剂量和活性标准。因此,临床II期参考意义相对有限。
事实上,根据2017年12月颁布的《细胞制品研究与评价技术指导原则(试行)》,明确我国细胞治疗临床试验采用“早期临床试验阶段”和“确证性临床试验阶段”两个阶段的形式。
早期临床试验阶段的研究内容原则上应包括初步的安全性评价、药代动力学研究、初步的药效学研究和剂量探索研究。并指出“必要时鼓励与药品审评机构沟通交流,以确保确证性临床试验方案设计的合理性,有利于研究结果的研判和拟申报产品的注册上市”。
《原则》解读中明确“非注册临床实验数据的可接受程度取决于临床试验用样品与申报注册产品的一致性以及临床研究数据的产生过程,数据的真实性、完整性、准确性和可溯源性,以及国家食品药品监督管理总局对临床试验的核查结果等情况综合科学评价。”
在国内企业中,金斯瑞生物科技(南京传奇)、安科生物(博生吉安科)、上海优卡迪、北京艺妙神州、深圳因诺免疫等均有不同程度的非注册临床数据。注册临床试验进程及商业化进程有望超出市场预期。
- 分享到:


